
AI-Accelerated Drug Design & Discovery
Drawing from my prior research experience in quantum chemistry and materials physics, I believe that integrating insights from physics into deep learning architectures will offer a potent complementary approach towards solving long-standing challenges in drug discovery (e.g., high-throughput quantum mechanical-level molecular dynamics simulations). The good news is, we have seen more recent SOTA models drawing inspiration from fundamental physics concepts, e.g., the principles behind diffusion models can be traced back to non-equilibrium thermodynamics. Below are a few relevant drug discovery projects that I have been working on.
Exploring Protein Conformation Space with Diffusion Generative Modeling
In the intricate realm of biochemistry, the distribution of protein 3D conformations (i.e., the Boltzmann distribution) plays a pivotal role in dictating cellular behavior and function. However, comprehending the vast conformational space of these proteins remains a challenging endeavor.
Leveraging the power of diffusion generative modeling, protein language models, folding models, and molecular dynamics force fields, our on-going project ventures into the potential energy landscape of protein conformations.
We aim to provide a more holistic and detailed understanding of protein thermodynamic properties as well as the transition pathways between different functional states. Such insights are crucial for predicting protein behaviors in varying conditions, and designing therapeutic strategies in drug discovery.




Animation | Sampling protein conformations through a diffusion process.
Learning Harmonic Molecular Surface Representations
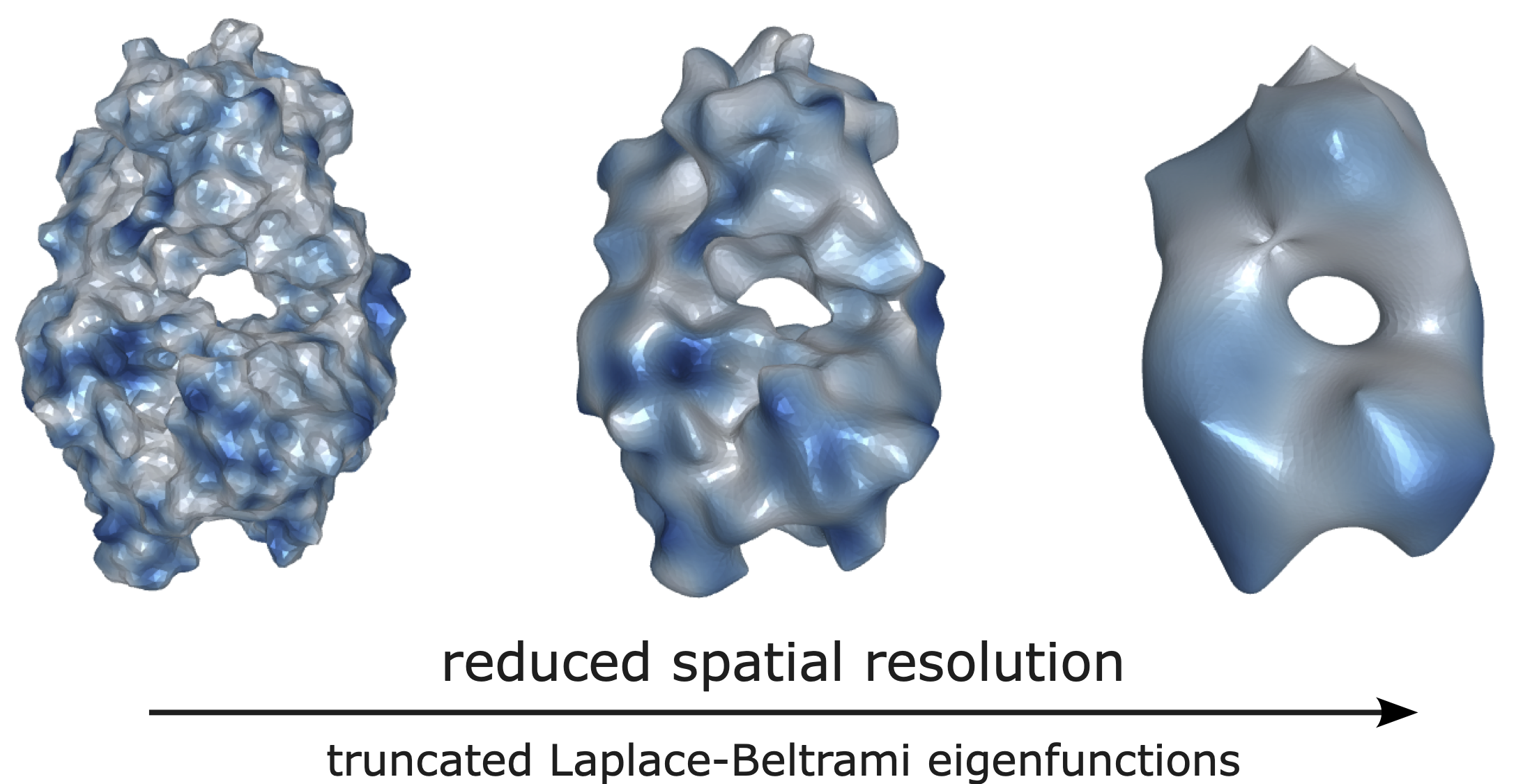
Our objective here is to properly encode the chemical and geometric information of protein systems for downstream applications, e.g., predicting whether an antibody could bind with a virus. However, protein-protein interactions are governed by sophisticated physical rules (e.g., hydrogen bond, hydrophobic interactions, etc.), hindering high-throughput screening of active reagents. Our proposed solution is based on a simple analogy:
Predicting protein-protein interactions is akin to piecing together a puzzle!
We argue that the necessary and sufficient condition for two proteins to interact is that they exhibit matching geometry (i.e., shape complementarity) as well as energetically favorable chemical patterns (e.g., opposite charge, similar hydrophobicity, etc.).
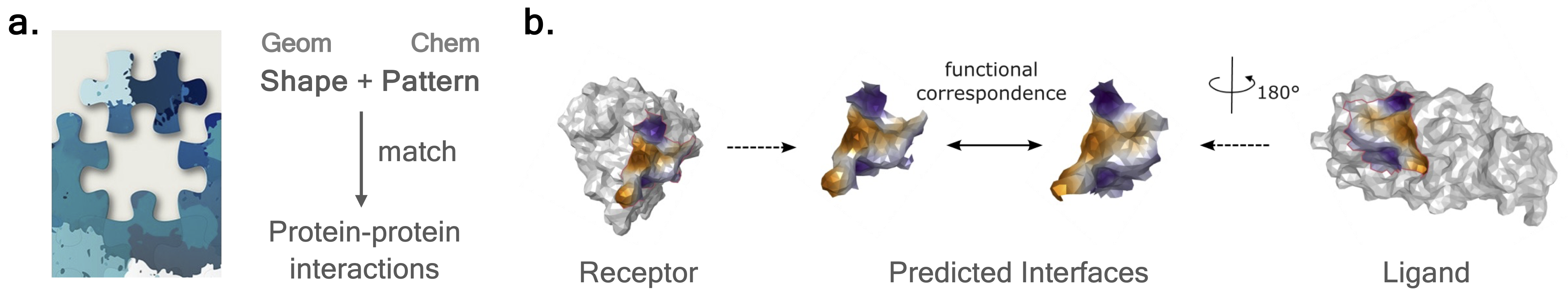
Figure | a. Shape complementarity and chemical affinity co-determine protein-protein interactions, just like piecing together a puzzle. b. We show that protein-protein interfaces indeed exhibit matching geometric and chemical patterns from the molecular surface perspective.
Following this idea, we propose a representation learning method to train a neural network to encode both geometric and chemical information of a protein surface. Specifically, the molecular surface is represented as a 2D Riemannian manifold, which adopts a set of Laplace-Beltrami eigenfunctions. The associated eigendecomposition is solely determined by the surface geometry, hence also getting the name "Shape DNA". Chemical attributes can be mapped as scalar functions on the manifold, which could alternatively be represented as the linear combination coefficients in its eigenspace.
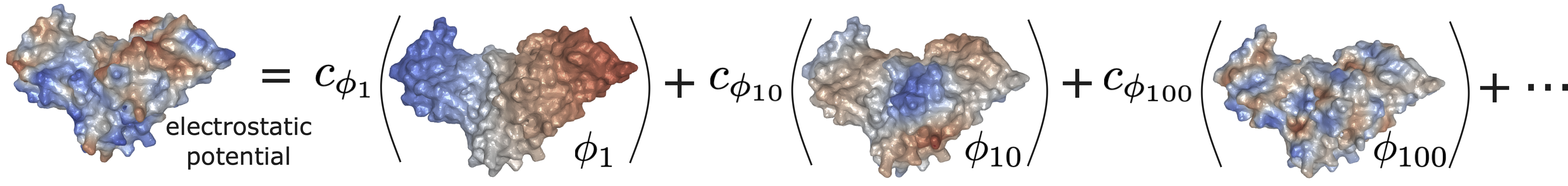
Figure | The electrostatic potential on a molecular surface manifold can be expanded as the linear combination of its Laplace-Beltrami eigenfunctions.
We then train a neural network to learn from known protein-protein interaction pairs and summarize their common patterns. Intuitively, our model learns to extract relevant geometric and chemical features that are correlated with binding, which helps it predict a reasonable binding site and subsequently outputs a docked protein complex structure. Specifically we employ functional maps to establish interface correspondence and realize protein docking through rigid transformations. The network architecture is shown below. Please see here for more details.

Figure | Rigid protein docking pipeline.
Molecular Geometric Deep Learning

Figure | Schematic protein-ligand complex with learnable inter-/intra-molecular interactions. The objective is to efficiently design candidate drug molecules exhibiting desirable properties.